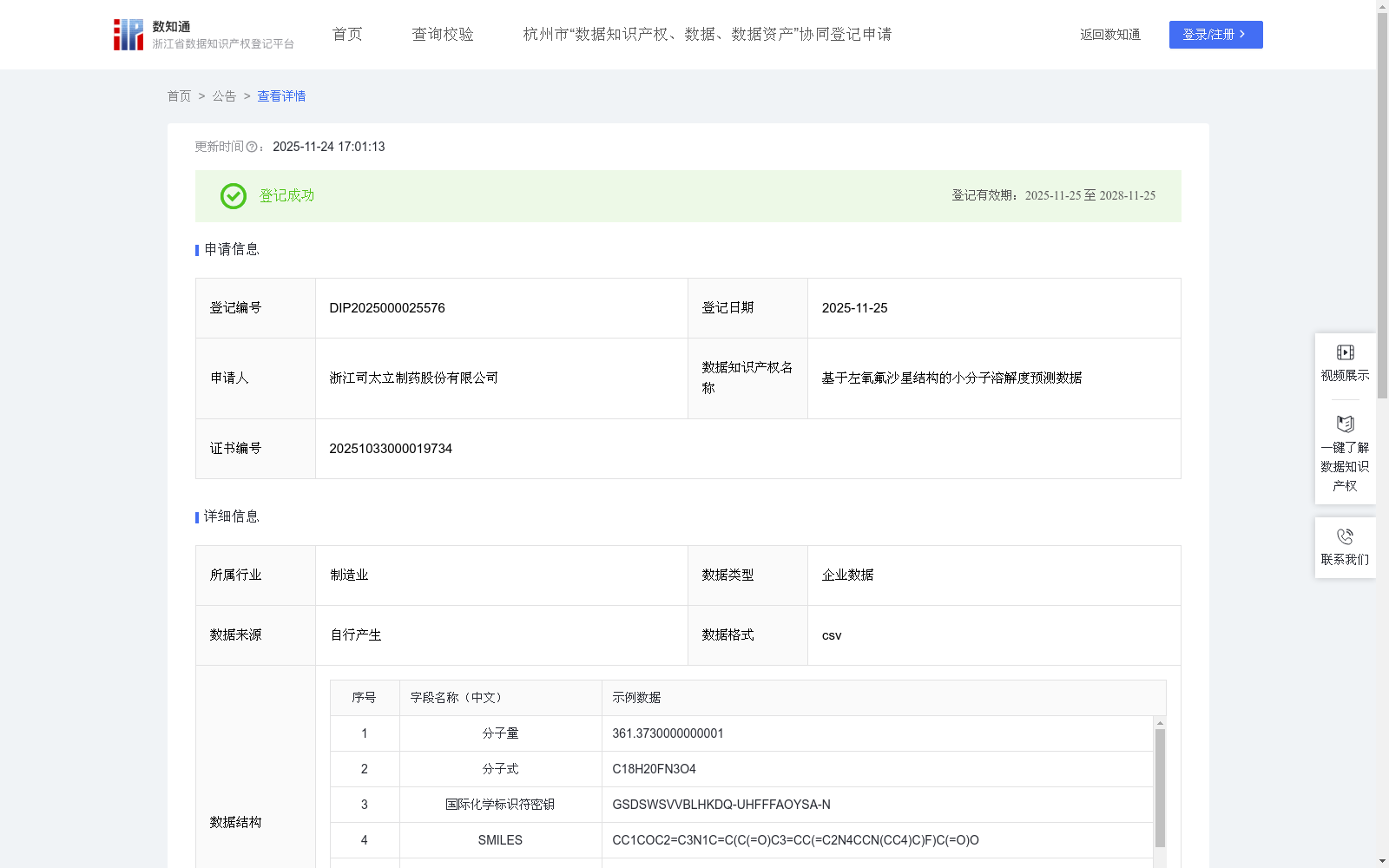
基于左氧氟沙星结构的小分子溶解度预测数据
收藏浙江省数据知识产权登记平台2025-11-24 更新2025-11-25 收录
下载链接:
https://www.zjip.org.cn/home/announce/trends/8405501
下载链接
链接失效反馈官方服务:
资源简介:
本数据的核心应用在于加速新型喹诺酮类药物及类似复杂多官能团分子的研发。
企业内部应用:在药物发现的早期阶段,研发团队可利用此数据训练的精准模型,对海量虚拟候选化合物进行高通量虚拟筛选,在无需化学合成的前提下,快速预测其水溶性。这能有效剔除溶解性差的分子,集中资源优化高潜力候选物,从而显著缩短新型喹诺酮类药物的研发周期并降低失败风险。此外,它还可用于指导已有先导化合物的结构修饰,以数据驱动的方式定向提升其成药性。
外部及行业应用:本数据集可作为化学信息学领域的专业基准,用于开发和验证针对多卤代、多羟基等复杂分子的新预测算法。同时,训练好的模型可以作为技术服务,授权给其他进入喹诺酮类领域的制药或化工企业,帮助其建立快速、低成本的分子溶解度评估能力,推动整个高端医疗材料行业的智能化发展。本研究基于支持向量回归(Support Vector Regression, SVR)算法构建了分子溶解度预测模型,该模型通过学习已知数据,实现对全新分子结构溶解度的预测。
1. 数据收集与特征加工:收集现有已知喹诺酮化合物的分子结构(以SMILES 字符串形式表示)、实验测定的溶解度对数值(logS),并采用166 位 MACCS(Molecular ACCess System)分子指纹作为特征表示,用于机器学习预测模型的构建。
2. 特征工程与模型构建
(1)特征工程:针对每个造影剂小分子(以左氧氟沙星类结构为代表)的 SMILES 字符串,通过 MACCS 结构密钥算法将其转换为166 位二进制特征向量(即 MACCS 指纹),作为模型的输入特征矩阵X;以对应的(logS)实验值作为模型的预测目标(输出变量Y)。
(2)模型构建:采用支持向量回归(SVR)算法构建预测模型,核心参数与结构如下:核函数:选用非线性径向基函数(Radial Basis Function, RBF),以适配分子特征与溶解度之间的复杂非线性关系;关键超参数:正则化参数C设为2.0(平衡模型拟合能力与泛化能力),核系数(gamma)设为(0.01)(控制径向基函数的局部影响范围);
(3)预测规则:模型训练完成后,生成623个支持向量及其对偶系数(权重),并得到截距项。对于新分子,其溶解度预测值通过如下方式计算:溶解度预测值=支持向量与新分子 MACCS 指纹的相似度加权和+截距(其中 “相似度” 由 RBF 核函数定义,加权和由支持向量的对偶系数决定)。
3. 预测结果的分类
判定规则模型直接输出的溶解度预测值为连续型浮点数。
为便于成药性评估与决策,将预测值进一步转化为分类标记,规则如下:若溶解度预测值> -4.0,标记为“高溶解度”,提示该分子成药潜力较高;若溶解度预测值在[-5.5, -4.0]范围内,标记为“中等溶解度”,提示分子性质可接受,可作为结构优化的候选对象;若溶解度预测值 < -5.5,标记为 “溶解性差”,提示该分子存在较高的成药性风险。
The core application of this dataset is to accelerate the research and development of novel quinolone drugs and similar complex multifunctional molecules.
Internal enterprise applications: In the early stage of drug discovery, R&D teams can use the precise models trained on this dataset to conduct high-throughput virtual screening on massive virtual candidate compounds, and rapidly predict their aqueous solubility without the need for chemical synthesis. This effectively eliminates molecules with poor solubility, concentrates resources on optimizing high-potential candidates, thereby significantly shortening the R&D cycle of novel quinolone drugs and reducing failure risks. Additionally, it can be used to guide structural modification of existing lead compounds, and directionally improve their druggability in a data-driven manner.
External and industry applications: This dataset can serve as a professional benchmark in the field of cheminformatics, used to develop and verify new prediction algorithms for complex molecules such as polyhalogenated and polyhydroxyl ones. Meanwhile, the trained models can be provided as technical services and licensed to other pharmaceutical or chemical enterprises entering the quinolone field, helping them establish rapid and low-cost molecular solubility assessment capabilities, and promoting the intelligent development of the entire high-end medical materials industry. This study constructed a molecular solubility prediction model based on Support Vector Regression (SVR) algorithm, which realizes the prediction of the solubility of brand-new molecular structures by learning known data.
1. Data Collection and Feature Processing: Collect the molecular structures of existing known quinolone compounds (represented by SMILES strings) and experimentally determined logarithm of solubility (logS), and adopt 166-bit MACCS (Molecular ACCess System) molecular fingerprints as feature representations for the construction of machine learning prediction models.
2. Feature Engineering and Model Construction
(1) Feature Engineering: For the SMILES strings of each small molecule of contrast agent (represented by levofloxacin-like structures), convert them into 166-bit binary feature vectors (i.e., MACCS fingerprints) via the MACCS structural key algorithm, which serve as the input feature matrix X of the model; use the corresponding experimental (logS) values as the prediction target (output variable Y) of the model.
(2) Model Construction: A prediction model was constructed using the Support Vector Regression (SVR) algorithm, with core parameters and structure as follows: Kernel function: The nonlinear Radial Basis Function (RBF) is selected to adapt to the complex nonlinear relationship between molecular features and solubility; Key hyperparameters: The regularization parameter C is set to 2.0 (to balance the model's fitting ability and generalization ability), and the kernel coefficient (gamma) is set to 0.01 (to control the local influence range of the Radial Basis Function);
(3) Prediction Rules: After the model is trained, 623 support vectors and their dual coefficients (weights) are generated, and the intercept term is obtained. For new molecules, their solubility prediction value is calculated as follows: Solubility prediction value = Weighted sum of the similarity between support vectors and the MACCS fingerprints of new molecules + Intercept term, where the "similarity" is defined by the RBF kernel function, and the weighted sum is determined by the dual coefficients of the support vectors.
3. Classification of Prediction Results
Judgment Rules: The solubility prediction value directly output by the model is a continuous floating-point number.
To facilitate druggability assessment and decision-making, the prediction values are further converted into classification labels according to the following rules: If the solubility prediction value > -4.0, it is marked as "High Solubility", indicating that the molecule has relatively high drug development potential; If the solubility prediction value is within the range of [-5.5, -4.0], it is marked as "Medium Solubility", indicating that the molecular properties are acceptable and can be used as candidates for structural optimization; If the solubility prediction value < -5.5, it is marked as "Poor Solubility", indicating that the molecule has a relatively high risk of druggability.
提供机构:
浙江司太立制药股份有限公司创建时间:
2025-10-13
搜集汇总
数据集介绍
背景与挑战
背景概述
该数据集包含2315条基于左氧氟沙星结构的小分子溶解度预测数据,涵盖分子量、SMILES、MACCS指纹等12个结构特征字段,用于通过支持向量回归算法预测溶解度并分类为高、中、差等级。其核心应用是加速喹诺酮类药物的研发,通过虚拟筛选优化候选化合物,提升药物发现效率。
以上内容由遇见数据集搜集并总结生成


